# 加载原始的、有缺失值的数据集
data("PimaIndiansDiabetes2", package = "mlbench")
dim(PimaIndiansDiabetes2)
## [1] 768 9
str(PimaIndiansDiabetes2)
## 'data.frame': 768 obs. of 9 variables:
## $ pregnant: num 6 1 8 1 0 5 3 10 2 8 ...
## $ glucose : num 148 85 183 89 137 116 78 115 197 125 ...
## $ pressure: num 72 66 64 66 40 74 50 NA 70 96 ...
## $ triceps : num 35 29 NA 23 35 NA 32 NA 45 NA ...
## $ insulin : num NA NA NA 94 168 NA 88 NA 543 NA ...
## $ mass : num 33.6 26.6 23.3 28.1 43.1 25.6 31 35.3 30.5 NA ...
## $ pedigree: num 0.627 0.351 0.672 0.167 2.288 ...
## $ age : num 50 31 32 21 33 30 26 29 53 54 ...
## $ diabetes: Factor w/ 2 levels "neg","pos": 2 1 2 1 2 1 2 1 2 2 ...7 数据准备
本合集主要使用了三个示例数据,使用最多的是著名的皮马印第安人糖尿病数据集,除此之外还使用了R语言自带的鸢尾花数据集以及德国信用评分数据集。
这里主要为大家简单介绍下印第安人糖尿病数据集的情况。
皮马印第安人糖尿病数据集最初来自国家糖尿病、消化和肾脏疾病研究所,包含来自美国亚利桑那州凤凰城附近的768名女性的信息。检测结果为是否患有糖尿病,其中268人检测呈阳性,500人检测呈阴性。该数据集有8个预测变量:怀孕次数、OGTT(口服葡萄糖耐量试验)、血压、皮肤皱褶厚度、胰岛素、BMI、年龄、糖尿病谱系功能。自1965年以来,国家糖尿病、消化和肾脏疾病研究所每隔两年对皮马人群进行一次研究。由于流行病学证据表明T2DM是遗传和环境因素相互作用的结果,因此皮马印第安人糖尿病数据集包含的有关信息可能与糖尿病的发病及其未来的并发症有关。
印第安人糖尿病数据集是一个二分类数据,我们需要使用多种指标预测患者是否有糖尿病,原始的数据集有很多缺失值,我们需要对它进行插补,在正式开始使用之前,先探索一下这个数据集。
该数据集一共有768个样本,9个变量,其中diabetes是二分类的结果变量,因子型,其他变量都是预测变量,且都是数值型。
各变量意义解释:
pregnant:怀孕次数glucose:血浆葡萄糖浓度(葡萄糖耐量试验)pressure:舒张压(毫米汞柱)triceps:三头肌皮褶厚度(mm)insulin:2小时血清胰岛素(mu U/ml)mass:BMIpedigree:糖尿病谱系功能,是一种用于预测糖尿病发病风险的指标,该指标是基于家族史的糖尿病遗传风险因素的计算得出的。它计算了患者的家族成员是否患有糖尿病以及他们与患者的亲缘关系,从而得出一个综合评分,用于预测患糖尿病的概率。age:年龄diabetes:是否有糖尿病,pos表示有糖尿病,neg表示没有糖尿病
我们直接使用GGally包探索一下各个变量的分布情况以及和结果变量之间的关系:
library(GGally)
ggpairs(PimaIndiansDiabetes2,columns = c(1:8),
mapping=aes(color=diabetes,alpha=0.3),
upper = list(
continuous = wrap("cor", size = 2.5) # 改变correlation的font size
)
)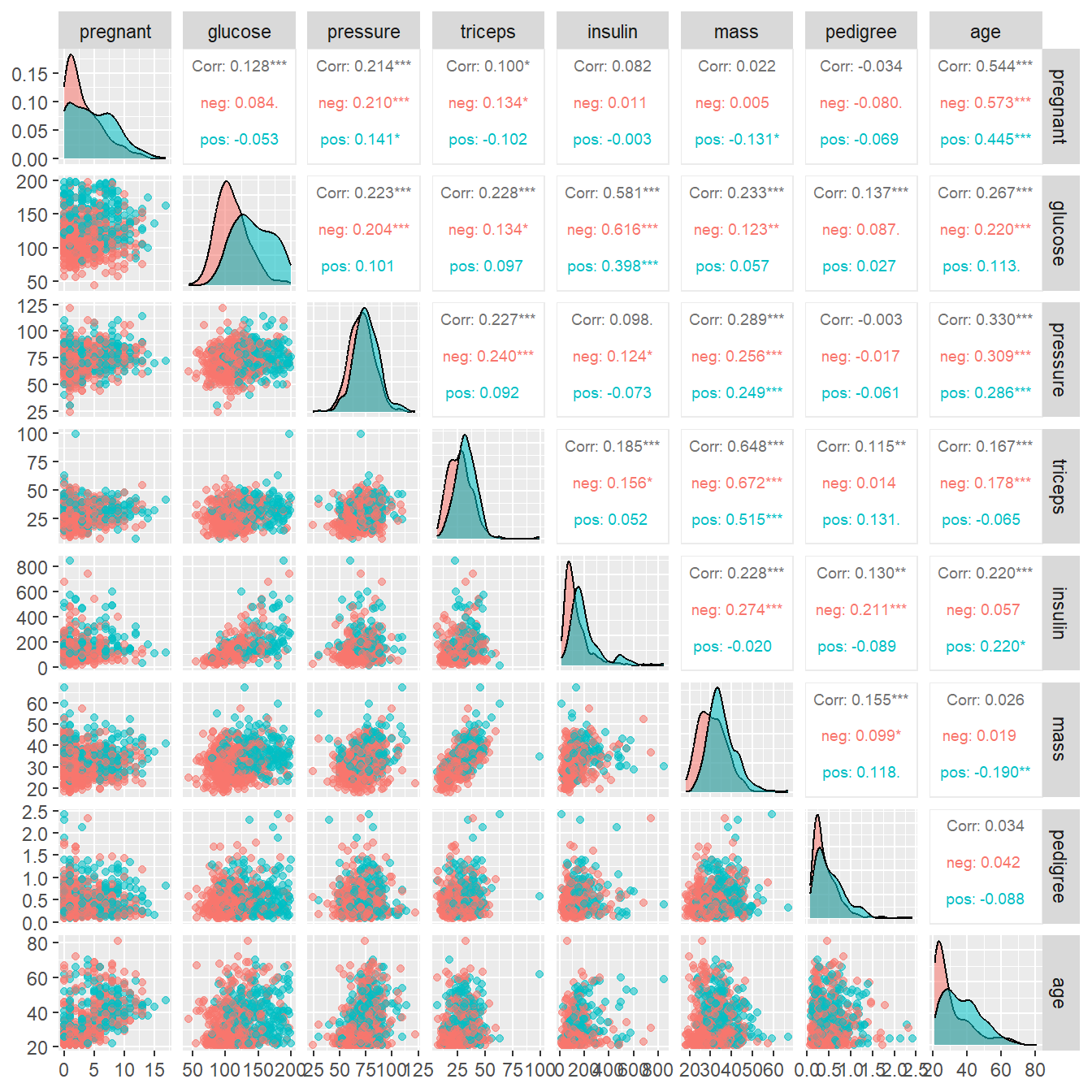
这张图其实就是一个相关系数矩阵的可视化,它展示了8个预测变量彼此之间的相关性。比如第一行第二列中的0.128是pregnant和glucose的相关系数。中间的密度曲线图展示了各个变量在不同组别(有糖尿病和没有糖尿病)的密度分布。
这里面有两个变量的相关性是比较高的,mass和triceps的相关性是0.648(其实还好,不算非常高)。
再简单查看下不同指标在糖尿病和非糖尿病组的分布有没有差异:
ggbivariate(PimaIndiansDiabetes2, outcome = "diabetes")+
scale_fill_brewer(type = "qual")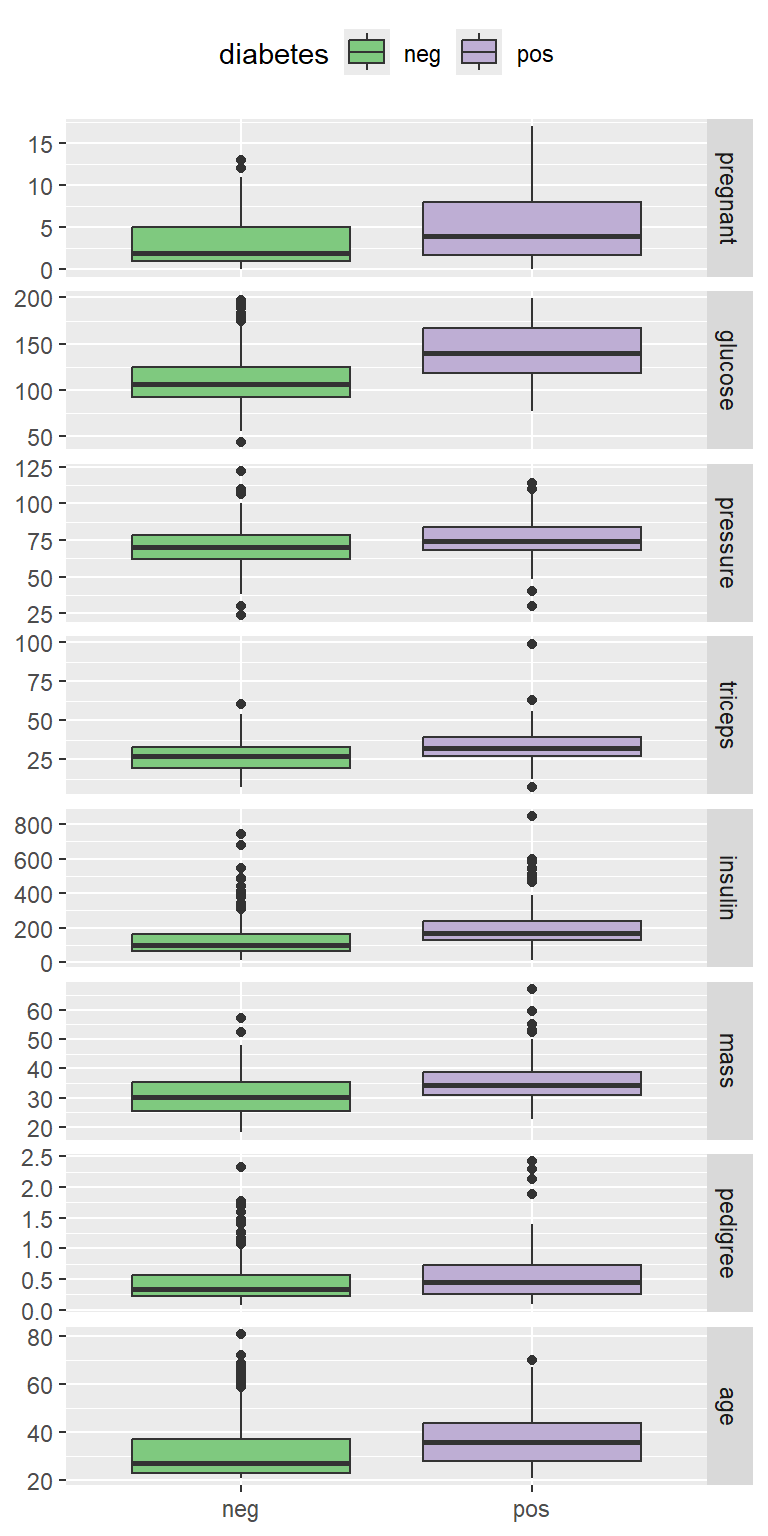
可以看出各个指标在不同组别确实是有点差异的哦。
下面简单探索下数据缺失情况:
library(visdat)
#vis_dat(PimaIndiansDiabetes2)
vis_miss(PimaIndiansDiabetes2)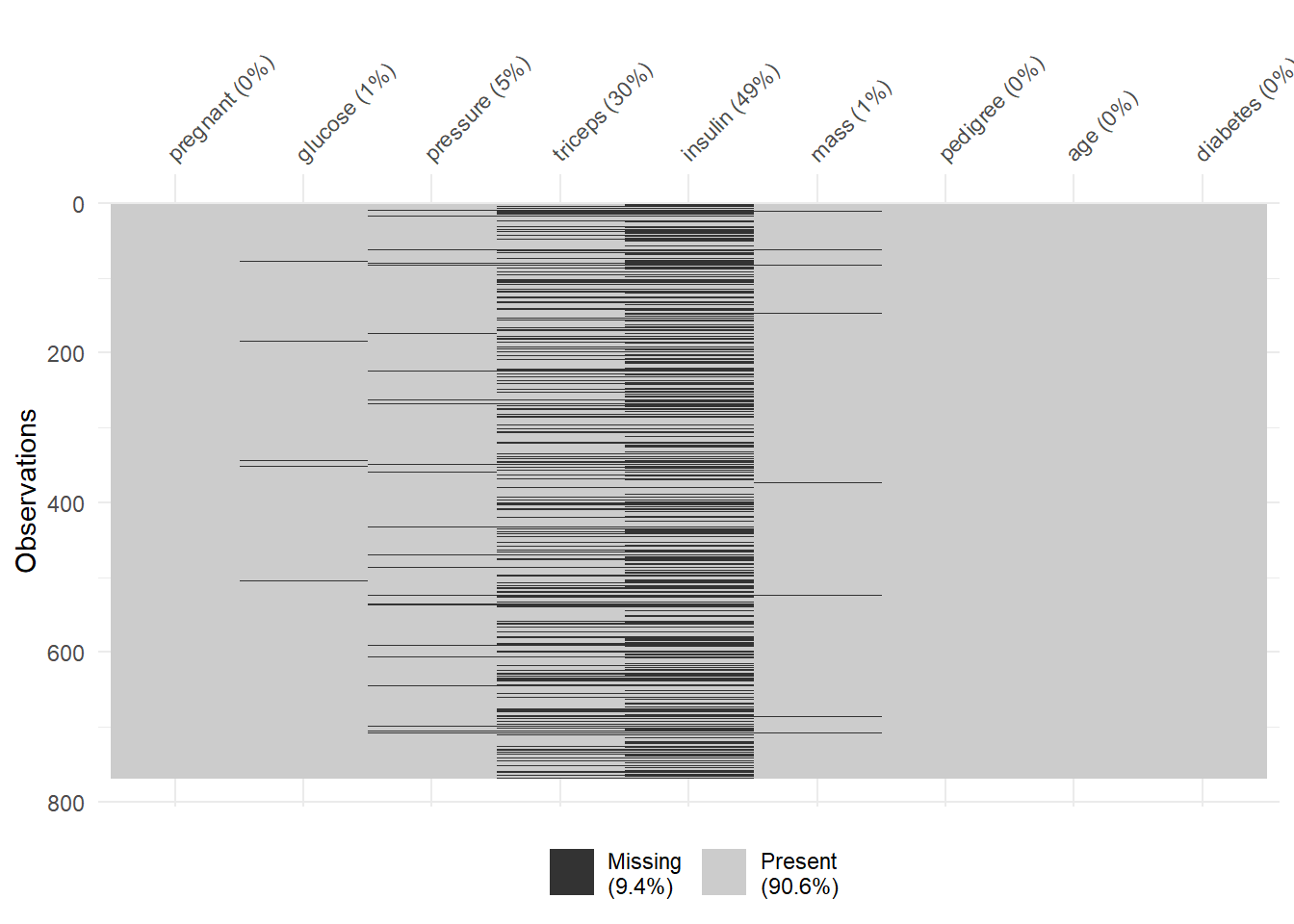
可以看到triceps和insulin这两列的缺失值比较多。
下面我们使用随机森林算法插补缺失值:
library(missForest)
set.seed(1234)
pimadiabetes <- missForest(PimaIndiansDiabetes2)$ximp
# 没有缺失值了
vis_miss(pimadiabetes)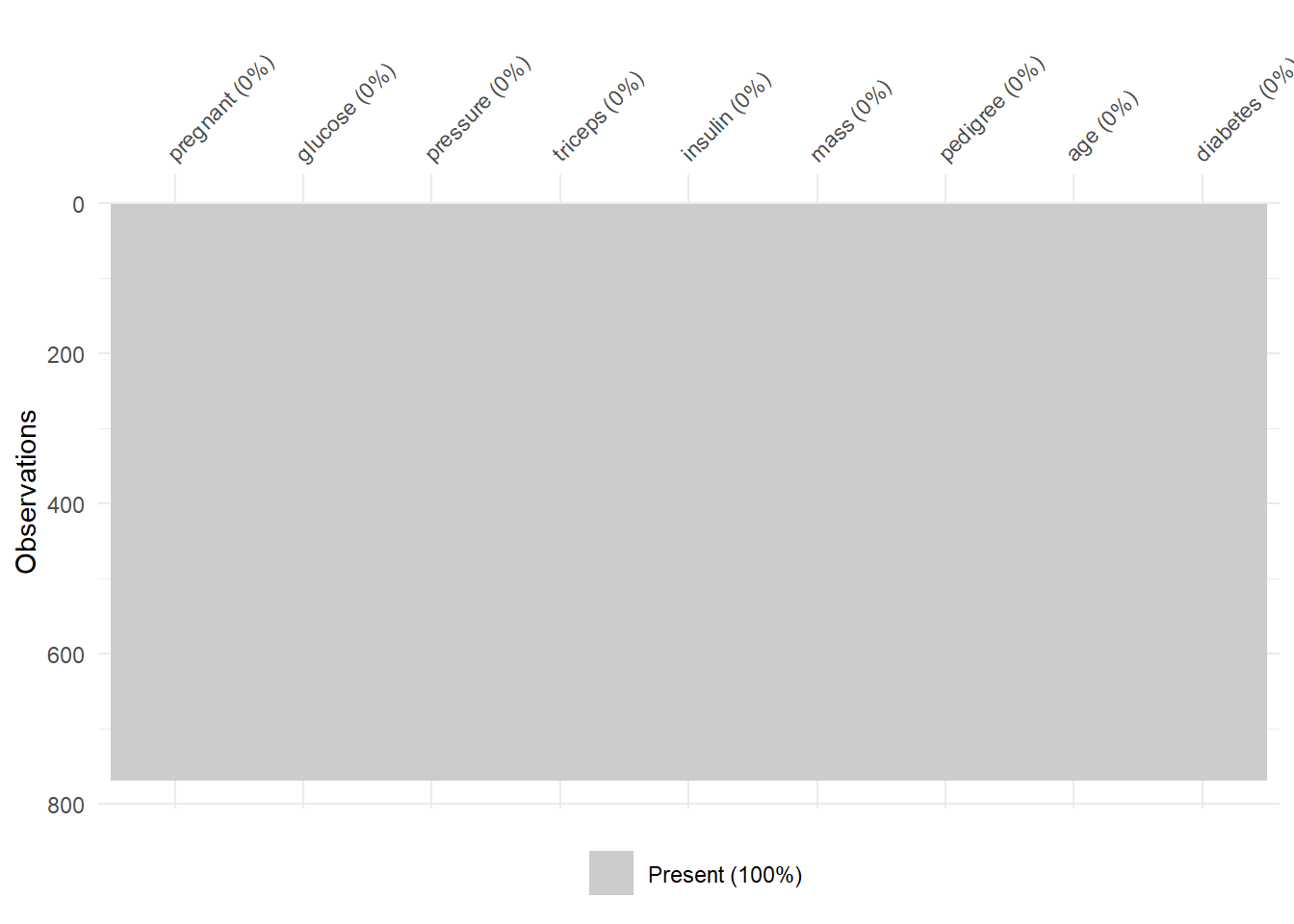
默认的结果变量diabetes是因子型的,但是它的因子顺序是先neg再pos,通常我们需要把阳性结果的顺序放在前面,所以这里我们修改一下:
levels(pimadiabetes$diabetes) <- c("pos","neg")
str(pimadiabetes)
## 'data.frame': 768 obs. of 9 variables:
## $ pregnant: num 6 1 8 1 0 5 3 10 2 8 ...
## $ glucose : num 148 85 183 89 137 116 78 115 197 125 ...
## $ pressure: num 72 66 64 66 40 ...
## $ triceps : num 35 29 22.9 23 35 ...
## $ insulin : num 202.2 64.6 217.1 94 168 ...
## $ mass : num 33.6 26.6 23.3 28.1 43.1 ...
## $ pedigree: num 0.627 0.351 0.672 0.167 2.288 ...
## $ age : num 50 31 32 21 33 30 26 29 53 54 ...
## $ diabetes: Factor w/ 2 levels "pos","neg": 2 1 2 1 2 1 2 1 2 2 ...保存下来方便后续使用:
save(pimadiabetes, file = "datasets/pimadiabetes.rdata")后续的分类任务演示都是使用的pimadiabetes这个数据集哦。